Risks and Complications
What are the risks and complications of surgeries for Apert syndrome?
All procedures for correcting individual deformities related to Apert syndrome carry a different set of risks and potential complications. Extensive craniofacial procedures tend to carry a higher risk profile, but are still practiced safely in designated craniofacial centers. Airway complications are the most serious issue encountered. Anesthesia pre-evaluation is necessary to stratify risk and to make provisions at the time of surgery for successful intubation and postoperative management. In general, many of the risks encountered during surgery for Apert syndrome are directly proportional to the severity of the condition.
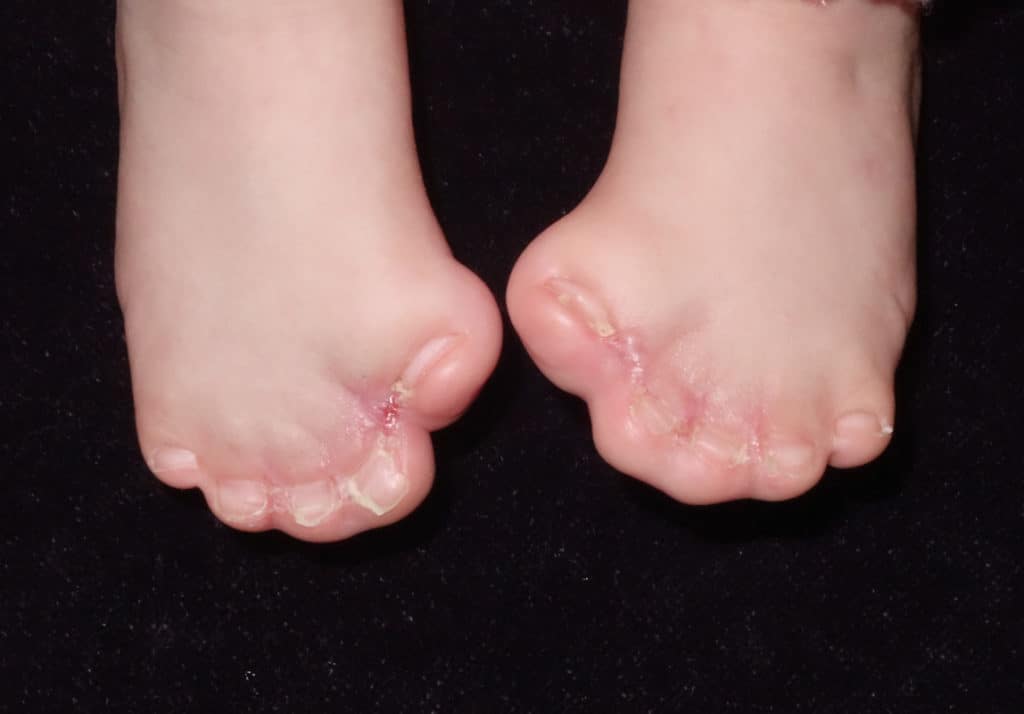
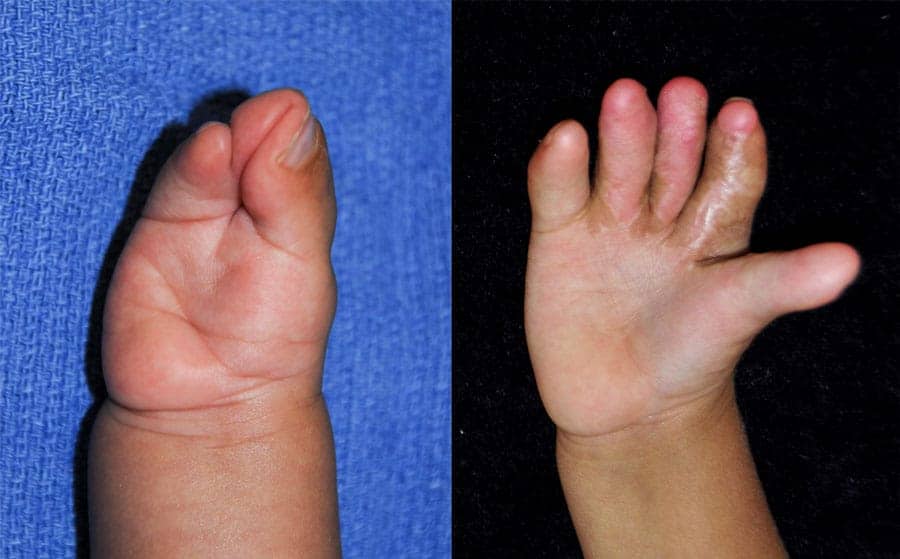
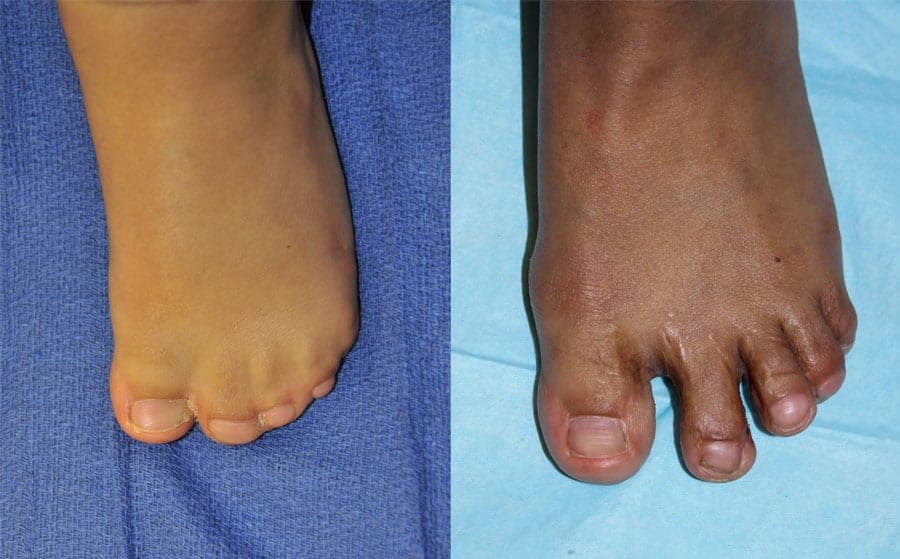
Stiffness of finger joints is common in Apert syndrome, with or without surgery. However, although fine finger movement is difficult to achieve, children with corrected Apert hand deformities lead very active lives with minimal limitations. When releasing webs, a chance of partial recurrence of the web is possible. This is called “webspace creep.” This sometimes requires deepening of spaces with another round of surgery or in combination with another procedure. Rarely, vascular compromise may lead to loss of a finger or toe. In addition, a rare complication of casting or compressive dressings is an elevation of pressures within compartments of the extremities, resulting in loss of circulation, nerve damage, and extreme pain. This requires emergent release of tight dressings and casts to avoid complications.
Bleeding, infection, and anesthetic complications are risks of any surgery. In addition, poor scarring or wound problems can occur rarely. Alopecia, or hair loss, may occur along the incision line within the scalp. More frequently, although improvement in function and appearance are possible, achieving perfect symmetry is elusive. Overcorrection or undercorrection may occur. Many times, revisional surgery will be required to improve scars, contour, or symmetry.